The unifying theme in my research is the development and use of novel computational methods to study macroevolutionary dynamics through time and space using large-scale datasets (“Big” data), with a particular focus on fossils and morphology. Fossils offer primarily (in most cases, exclusively) morphological data, and remain our only source of direct evidence of the true evolutionary history of life on Earth.
Currently, I am working on a project exploring morphological evolution of planktonic foraminifer communities across the Cretaceous-Paleogene (K-Pg) boundary using deep learning and computer vision. This project encompasses the major focal points of my research program, which are:



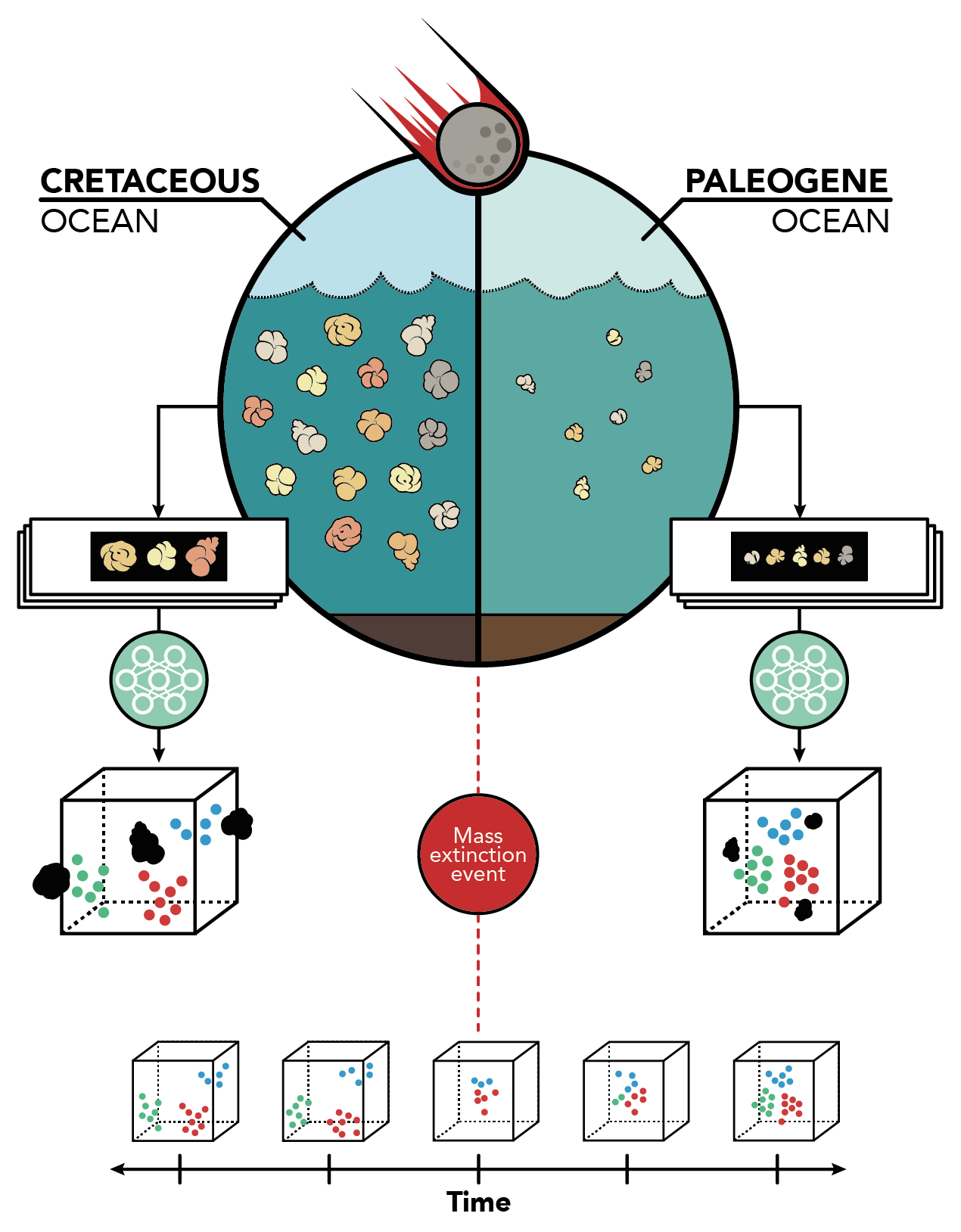
The K-Pg mass extinction occurred ~66 million years ago when a large meteorite collided with the Earth, famously leading to the extinction of the dinosaurs. Other organisms were also dramatically affected by the event – for instance, it is estimated that >90% of planktonic foraminifer species went extinct at this time. As planktonic foraminifera are important contributors to the biogeochemical cycles of the ocean (contributing up to 56% and 80% of global oceanic carbonate export and deep-marine calcite, respectively), this sudden mass extinction lead to a severely disrupted biological pump, decreased productivity, and a large restructuring of the ocean ecosystem. Studying the evolution of planktonic foraminifera across the K-Pg boundary thus grants us a vital view into how ocean dynamics change and respond to large-scale perturbations. Studying these effects is especially important as anthropogenic climate change continues to rapidly and dramatically disrupt ecosystems in our modern world.
In this project, a high-resolution morphological record of planktonic foraminifera across the K/Pg boundary will be generated using high-throughput imaging (facilitated by a Keyence VHX-7000 4K high-resolution microscope) and deep learning. This will result in a Big dataset that will allow us to study the morphological and ecological evolution of planktonic foraminifer communities across the boundary, as well as elucidate how these processes correlate with geophysical and geochemical systems in the face of global catastrophe.
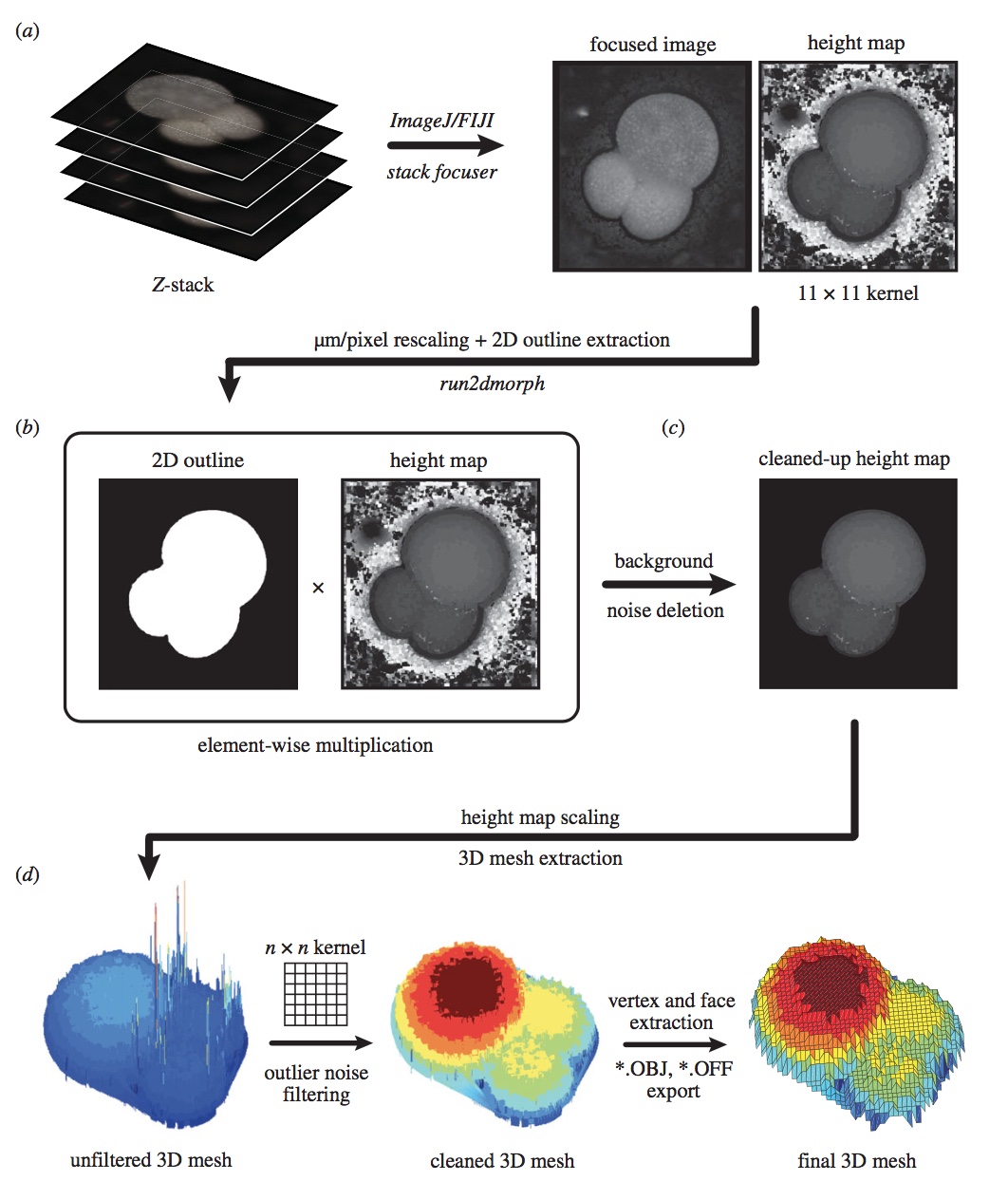 One of the major hindrances to the collection of large-scale morphological (“phenomic”) datasets is the sheer amount of time and hand labor required to identify and measure morphological features across a large number of taxa. To tackle this issue, I work on developing methods for automatically extracting morphological information from high-throughput images of both extant and fossil organisms. This includes AutoMorph, an open source software package for automatically extracting 2D and 3D data from focus-stacked images. Software like AutoMorph allows for the collection of community-scale morphological data, which can then be used to understand interactions among macroevolutionary and macroecological processes, population-level variability, evolutionary dynamics across biogeographic space, etc.
One of the major hindrances to the collection of large-scale morphological (“phenomic”) datasets is the sheer amount of time and hand labor required to identify and measure morphological features across a large number of taxa. To tackle this issue, I work on developing methods for automatically extracting morphological information from high-throughput images of both extant and fossil organisms. This includes AutoMorph, an open source software package for automatically extracting 2D and 3D data from focus-stacked images. Software like AutoMorph allows for the collection of community-scale morphological data, which can then be used to understand interactions among macroevolutionary and macroecological processes, population-level variability, evolutionary dynamics across biogeographic space, etc.
While AutoMorph performs well for datasets that are carefully prepared to reduce the amount of background noise and increase contrast between the specimens and the substrate, it can run into trouble when processing samples that do not follow these parameters. This is because AutoMorph uses traditional image processing methods such as RGB filters and contrast thresholding. To facilitate segmentation and processing of more difficult samples, I am also developing methods using state-of-the-art mask R-CNNs. This includes work done in collaboration with Nadia Irwanto (as her senior thesis in Computer Science at Yale University), Pincelli Hull (Yale University), and Elizabeth Sibert (Yale University).
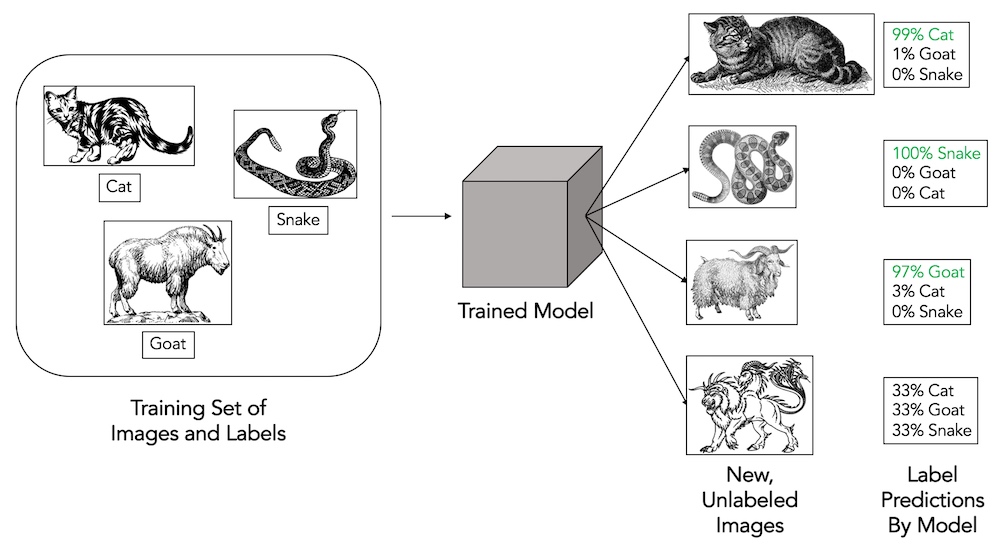 Machine learning and computer vision are important new technologies that we are beginning to bring fully to bear on applications in evolutionary biology. In light of the development of ever more sophisticated and accurate models for intelligent object and context recognition, we are applying these methods to the automatic identification of organisms in high-throughput images. Once trained, these models can provide taxonomic identifications for datasets containing hundreds of thousands of specimens – which would normally take months to years of work – in a matter of minutes to hours.
Machine learning and computer vision are important new technologies that we are beginning to bring fully to bear on applications in evolutionary biology. In light of the development of ever more sophisticated and accurate models for intelligent object and context recognition, we are applying these methods to the automatic identification of organisms in high-throughput images. Once trained, these models can provide taxonomic identifications for datasets containing hundreds of thousands of specimens – which would normally take months to years of work – in a matter of minutes to hours.
We are using deep learning and convolutional neural networks to train models that can automatically identify species of planktonic foraminifera with accuracies higher than those observed in human taxonomic experts. We compare in detail the results of human vs. machine classifiers, and find that using automated machine methods can avoid biases that human experts are susceptible to (such as the tendency to mistakenly classify rare species are more common species). The Endless Forams database and associated publication represent our significant initial contribution to this burgeoning field of study.
 The combination of high-throughput imaging/data extraction and automated taxonomy using deep learning opens up the possibility of generating large datasets that would allow us to study the evolution of entire communities across the globe and in deep time. For instance, the K-Pg CNN project aims to generate a dataset of over 600,000 individual specimens across 2.5 million years of biological history to study how foraminifer communities changed in response to the meteorite impact and recovered in the aftermath of mass extinction. Such datasets would not be feasible to create manually. The methods I work on thus greatly extend the breadth and depth of the types of questions we are able to answer.
The combination of high-throughput imaging/data extraction and automated taxonomy using deep learning opens up the possibility of generating large datasets that would allow us to study the evolution of entire communities across the globe and in deep time. For instance, the K-Pg CNN project aims to generate a dataset of over 600,000 individual specimens across 2.5 million years of biological history to study how foraminifer communities changed in response to the meteorite impact and recovered in the aftermath of mass extinction. Such datasets would not be feasible to create manually. The methods I work on thus greatly extend the breadth and depth of the types of questions we are able to answer.
In addition to the K-Pg project, we are also currently comparing the Endless Forams dataset with a second dataset sampled from the same underlying community (23 oceanic core sites that form a latitudinal transect across the Atlantic Ocean) that has species labels assigned using our trained supervised machine learning classifier. By running the same analyses to characterize community ecology on both the human-labeled dataset and the machine-labeled dataset, we demonstrate that the machine-lead approach is both accurate and feasible, and in fact avoids biases (with respect to specimen size and rarity) that the human dataset exhibits, thus avoiding the potential reconstruction of misleading biodiversity patterns.

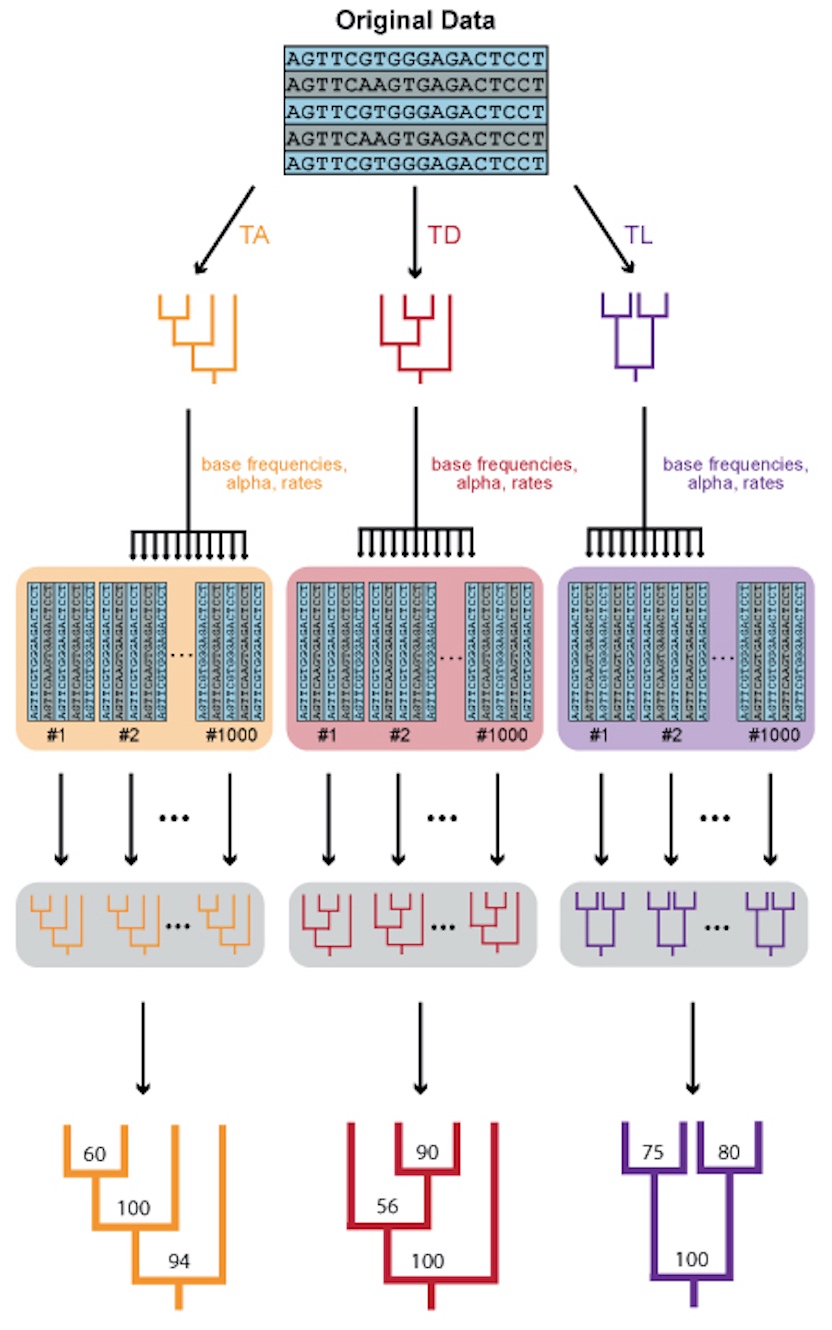 Phylogenies serve as a foundation for understanding evolutionary history and macroevolutionary processes and dynamics. As such, it is vital that the phylogenetic inference methods we use are as robust as possible. Because it is impossible to directly observe deep evolutionary history, congruence between phylogenies inferred using independent datasets is the only measure by which we can assess the “truth” of a given phylogenetic hypothesis. However, in practice, phylogenetic incongruence is pervasive, particularly between topologies inferred from genetic vs. phenotypic data.
Phylogenies serve as a foundation for understanding evolutionary history and macroevolutionary processes and dynamics. As such, it is vital that the phylogenetic inference methods we use are as robust as possible. Because it is impossible to directly observe deep evolutionary history, congruence between phylogenies inferred using independent datasets is the only measure by which we can assess the “truth” of a given phylogenetic hypothesis. However, in practice, phylogenetic incongruence is pervasive, particularly between topologies inferred from genetic vs. phenotypic data.
I studied phylogenetic incongruence by investigating possible sources of error and bias in phylogenomic inference using Bayesian and Maximum Likelihood frameworks, using the primary test case of the position of turtles within Amniota. Using simulations, statistical tests, data filtering, and gene/species tree comparisons, I explored sources of misleading signal and systematic biases, model misspecification, and the consequences of deep coalescence when inferring phylogenies in deep time. This work comprises my Ph.D. thesis, which was completed at Yale University under the supervision of Jacques Gauthier.
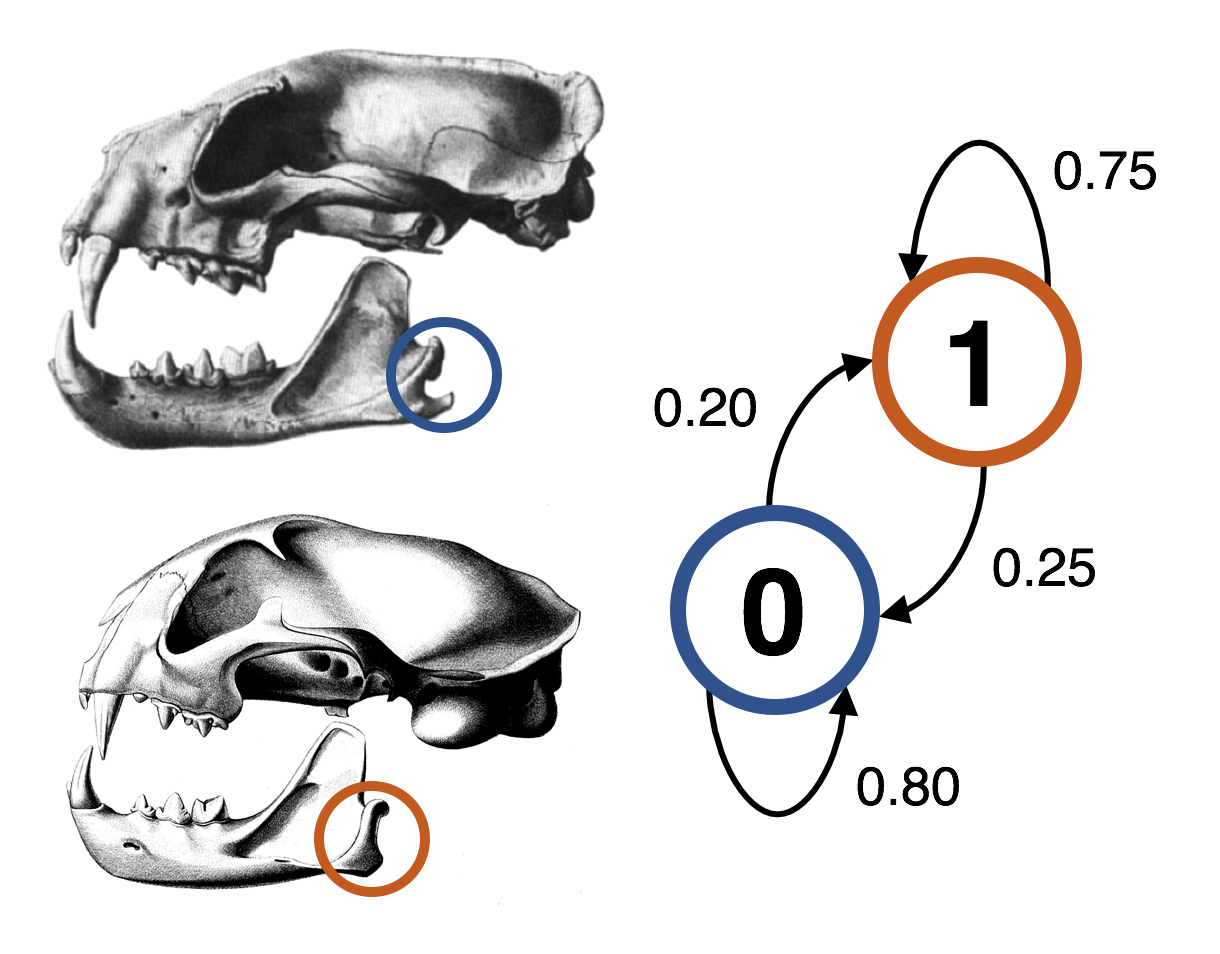 Although many statistical models for molecular evolution within a phylogenetic framework exist, development of analogous models of morphological evolution has lagged behind. This is largely due to the complexity of morphological evolution; for instance, form and structure can be affected by difficult-to-constrain confounding phenomena such as homoplasy or physical forces. Furthermore, unique difficulties exist for morphological character coding, both practical (e.g., 0’s and 1’s are not comparable across characters the way that A’s, G’s, C’s, and T’s are across sites) and theoretical (e.g., hypotheses of primary homology remain, to some degree, subjective).
Although many statistical models for molecular evolution within a phylogenetic framework exist, development of analogous models of morphological evolution has lagged behind. This is largely due to the complexity of morphological evolution; for instance, form and structure can be affected by difficult-to-constrain confounding phenomena such as homoplasy or physical forces. Furthermore, unique difficulties exist for morphological character coding, both practical (e.g., 0’s and 1’s are not comparable across characters the way that A’s, G’s, C’s, and T’s are across sites) and theoretical (e.g., hypotheses of primary homology remain, to some degree, subjective).
The lack of sophisticated statistical models describing morphological evolution hinders the use of morphology in modern phylogenetic inference and comparative method frameworks. To address this, I am working on developing new probabilistic models of morphological evolution, including models to capture correlation structure in large phenomic datasets in order to automatically cluster characters into coevolving modules. This work is done in collaboration with Fredrik Ronquist (Swedish Museum of Natural History).